These susceptible isles
October 7, 2024
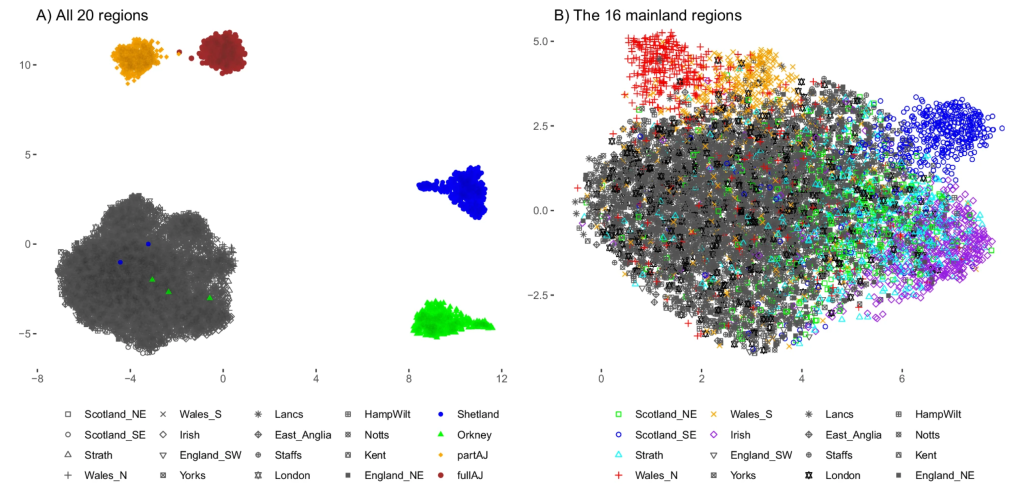
Fascinating insights into the regional differentiation within the UK and Ireland – with Mike Halachev, Jim Wilson and others. Remarkably, even with the current – far from complete – genomic coverage of regional populations we see evidence for different rare variants within different regions. With conservative filtering we find that a fraction of these regionally enriched rare variants are likely to have adverse biomedical consequences in homozygous individuals. Now available on Nature Comms with commentary in the Economist – following a marathon review process of ~2 years!